Modifying an existing Compocyte classifier.¶
In the previous tutorial, we labelled a PBMC dataset, fit an explicitly defined label (and thus classifier) hierarchy to our labels and trained a Compocyte classifier to assign cell type labels to new data. Now, we want to take a look at the ways in which a hierarchical classifier permits changes in a way a monolithic classifier would not.
We will start by loading the classifier we have previously trained.
[1]:
from Compocyte.core.hierarchical_classifier import HierarchicalClassifier
import scanpy as sc
# Load the trained classifier by specifying a save_path and calling the load() method.
# The save_path should be the same as the one used during training.
classifier = HierarchicalClassifier(
save_path="./exclude/pbmc_classifier"
)
classifier.load()
Neither graph nor dict_of_cell_relations defined upon initialization.
Please run .load() to load an existing classifier.
/usr/local/lib/python3.14/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
[2]:
test_adata = sc.read_h5ad("./exclude/test_adata.h5ad")
We already know that we get predictions when running our classifier on this data, but let’s see if they are any good.
[3]:
classifier.load_adata(test_adata)
classifier.predict_all_child_nodes('Blood')
Predicting at Blood.
Predicting at Lymphoid.
Predicting at T cells.
Predicting at Myeloid.
[4]:
sc.pl.dotplot(
classifier.adata,
var_names=['CD3D', 'CD4', 'CD8A', 'KLRB1', 'NCAM1', 'FCGR3A', 'CD19', 'CD38', 'CD14', 'VCAN', 'FCER1A', 'CLEC4C', 'HBB', 'ITGB3'],
groupby='Level_2_pred'
)
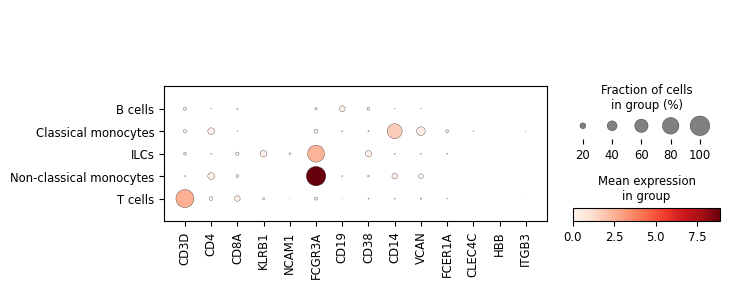
Not so bad for a classifier trained on 2000 cells. Let’s assume that I am quite interested in the B cell subset. However, I did not have sufficient data to train the classifier beyond the B cell label. But someone else did. We will now take the B cell branch of our the official pretrained PBMC classifier and attach it to the classifier we have trained here and see how it fares.
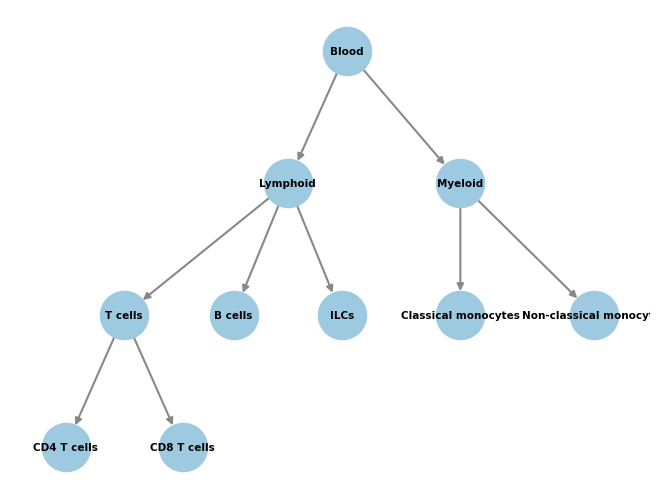
This is our classifier before expanding.
[5]:
import networkx as nx
from networkx.drawing.nx_agraph import graphviz_layout
# Plot the graph structure we gave to the classifier during training.
pos = graphviz_layout(
classifier.graph,prog="dot",
root='Blood',
args='-Gsplines=curved -Gnodesep=8 -Goverlap=scalexy -Gbeautify=false'
)
nx.draw(
classifier.graph, pos,
with_labels=True,
node_color="#9ecae1",
node_size=1200,
edge_color="#888",
width=1.5,
font_size=7.5,
font_weight="bold",
)
[6]:
from Compocyte.pretrained import pbmc_pretrained
pretrained_classifier = pbmc_pretrained()
Neither graph nor dict_of_cell_relations defined upon initialization.
Please run .load() to load an existing classifier.
[7]:
exported_pretrained = pretrained_classifier.export_classifiers("./exclude/exported_classifiers/")
exported_3k = classifier.export_classifiers("./exclude/exported_classifiers_3k/")
[8]:
pretrained_classifier.dict_of_cell_relations
[8]:
{'Blood': {'PL': {},
'ERY': {},
'MCP': {},
'leukocyte': {'PB': {'B': {'B-naive': {},
'B-memory': {'B-memory-DN': {},
'B-memory-switched': {},
'B-memory-non-switched': {}}},
'plasma-blast': {'plasma-blast_proliferating': {},
'plasma-blast_IgM': {},
'plasma-blast_IgG': {},
'plasma-blast_IgA': {}}},
'M': {'gran': {'baso': {}, 'neutro': {}},
'DC': {'AS-DC': {},
'cDC': {'cDC1': {}, 'cDC2': {}, 'cDC3': {}},
'p-DC': {}},
'mono': {'i-mono': {'i-mono_IFN-I': {}},
'mono_IFN-I': {},
'c-mono': {'c-mono_IFN-I': {},
'inf-c-mono': {},
'c-mono_inflammasome': {}},
'nc-mono': {'nc-mono_IFN-I': {}, 'nc-mono_inflammasome': {}}}},
'TNK': {'TNK_proliferating': {},
'ILC': {'NK': {'NK_proliferating': {},
'CD56dim-NK': {},
'CD56bright-NK': {},
'NK-adaptive': {}},
'ILC2': {}},
'T': {'T_proliferating': {},
'abT': {'abT_proliferating': {},
'NKT': {},
'CD4-T': {'CD4-T-naive': {},
'Treg': {'Treg_proliferating': {}, 'Treg_BATF': {}},
'CD4-TEM': {},
'CD4-TCM': {}},
'CD8-T': {'CD8-T-naive': {},
'CD8-TCM': {'CD8-TCM_preexhausted': {},
'CD8-TCM_terminal-exhaustion': {},
'CD8-TCM_nonexhausted': {}},
'CD8-T-effector': {'CD8-T-KLRG1neg-effector': {'CD8-T-KLRG1neg-effector_nonexhausted': {},
'CD8-T-KLRG1neg-effector_terminal-exhaustion': {},
'CD8-T-KLRG1neg-effector_preexhausted': {}},
'CD8-T-KLRG1pos-effector': {'CD8-T-KLRG1pos-effector_nonexhausted': {},
'CD8-T-KLRG1pos-effector_terminal-exhaustion': {},
'CD8-T-KLRG1pos-effector_preexhausted': {},
'CD8-T-KLRG1pos-effector_exhausted-progenitor': {}}}},
'TCM': {'TCM_proliferating': {}, 'TCM_nonexhausted': {}},
'T-naive': {},
'MAIT': {}},
'gdT': {}}}}}}
[9]:
exported_3k['Blood']['Lymphoid']['B'] = exported_pretrained['Blood']['leukocyte']['PB']['B']
[10]:
# Remove duplicate B cell node from the new hierarchy to avoid confusion during merging.
del exported_3k['Blood']['Lymphoid']['B cells']
[11]:
test_adata = sc.read_h5ad("./exclude/test_adata.h5ad")
[12]:
merged_classifier = HierarchicalClassifier(
save_path="./exclude/merged_classifier",
dict_of_cell_relations=exported_3k,
adata=test_adata,
# the new hierarchy goes one level deeper than the original one, so we need to add the new level to the obs_names
obs_names=classifier.obs_names + ['Level_4'],
temp_paths=["./exclude/exported_classifiers/", "./exclude/exported_classifiers_3k/"],
root_node=classifier.root_node
)
# The label encoding is specified at the local classifier level.
# Because we renamed the B cell node, we need to tell the classifier to update the label encoding as well.
merged_classifier.rename(old_label='B cells', new_label='B', parent_label='Lymphoid')
/workspaces/Compocyte/src/Compocyte/core/base/data_base.py:122: UserWarning: You have supplied normalized, log-transformed data. Please ensure that this is intended and data is normalized to 10_000 counts per cell prior to log1p transformation.
warn('You have supplied normalized, log-transformed data. Please ensure that \
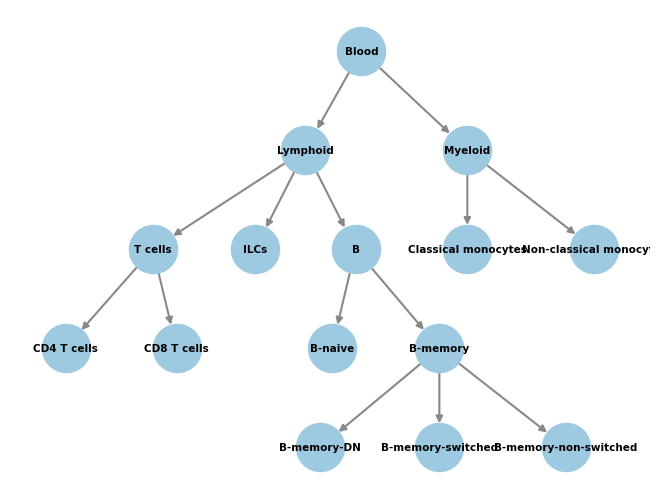
This is our classifier after expanding.
[13]:
# Plot the graph structure we gave to the classifier during training.
pos = graphviz_layout(
merged_classifier.graph,prog="dot",
root='Blood',
args='-Gsplines=curved -Gnodesep=8 -Goverlap=scalexy -Gbeautify=false'
)
nx.draw(
merged_classifier.graph, pos,
with_labels=True,
node_color="#9ecae1",
node_size=1200,
edge_color="#888",
width=1.5,
font_size=7.5,
font_weight="bold",
)
[14]:
merged_classifier.predict_all_child_nodes('Blood')
Predicting at Blood.
Predicting at Lymphoid.
Predicting at T cells.
Predicting at B.
Predicting at B-memory.
Predicting at Myeloid.
[15]:
sc.pl.dotplot(
merged_classifier.adata[merged_classifier.adata.obs.Level_2_pred == 'B'],
var_names=['CD19', 'CD38','MS4A1', 'CD27', 'TCL1A', 'TNFRSF13B'],
use_raw=True,
groupby='Level_3_pred'
)
/usr/local/lib/python3.14/site-packages/anndata/_core/anndata.py:1257: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.14/site-packages/anndata/_core/anndata.py:1257: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.14/site-packages/anndata/_core/anndata.py:1257: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.14/site-packages/anndata/_core/anndata.py:1257: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
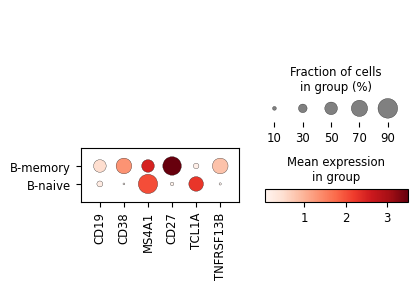
Would you look at that? With some Compocyte magic, we have found memory B cells that we could not have found without expanding the B cell classifier. Congratulations. If you would like to learn more have a look at our other tutorials.